Pencarian Regulatory Motif pada Bakteri TBC (Mycobacterium tuberculosis)
Artikel ini adalah upaya penyelesaian masalah yang ada di buku Bioinformatics Algorithm Vol 1 karya Pavel Pevzner dan Phillip Compeau, spesifiknya pada Chapter 2 tentang Finding Regulatory Motifs. Semua gambar dan materi oleh karenanya berasal dari buku tersebut.
Permasalahan Biologis: Kenapa bakteri TBC itu menyebalkan?
Bakteri tuberkulosis (Mycobacterium tuberculosis) adalah bakteri yang dapat menyebabkan penyakit TBC, sebuah penyakit mematikan yang pernah membuat seperempat penduduk eropa meninggal di abad ke-19. Meskipun kematian akibat bakteri ini sudah menurun signifikan akibat penemuan antibiotik, saat ini bakteri ini masih menjadi momok bagi banyak orang yang di setiap tahun dapat membunuh jutaan orang di bumi.
Alasan kenapa bakteri ini tetap berbahaya meskipun sudah ada antibiotik adalah karena kemampuan resistensinya terhadap antibiotik. Kapasitas ini diperoleh ketika bakteri TBC beralih dari keadaan aktif menjadi dalam keadaan dorman. Dalam keadaan dorman ini, bakteri TBC tak akan menunjukan aktivitasnya di dalam tubuh (disebut infeksi laten) tetapi dapat bertahan di dalam tubuh selama bertahun-tahun.
Salah satu pemicu utama yang membuat bakteri ini berubah dari keadaan aktif menjadi dorman adalah keadaan kekurangan oksigen atau hipoksia. Ketika bakteri kekurangan oksigen, salah satu protein faktor transkripsi bernama DosR (Dormancy Survival Regulator) akan mengatur ekspresi banyak gen sekaligus yang membuat bakteri tersebut menjadi dalam keadaan dorman. Dalam hal ini DosR perlu mengenali dan menempel pada sekuens pendek spesifik pada upstream region atau promoter dari gen-gen yang diregulasinya.
Dari pengetahuan tersebut, kita dapat membayangkan sebuah pertanyaan saintifik yang spesifik: “sekuens spesifik seperti apa di bagian promoter yang terdapat pada gen-gen tertentu yang aktif ketika proses dormasi yang dapat berikatan dengan protein DosR?”. Pola sekuens spesifik yang bekerja menjadi semacam sinyal untuk protein seperti DosR sendiri disebut sebagai Motif, sehingga pertanyaan saintifik itu akan berubah menjadi permasalahan mencari motif. Pertanyaannya sekarang adalah bagaimana cara untuk menyelesaikan permasalahan pencarian motif tersebut?
Penerjemahan Permasalahan Biologis menjadi Komputasional
Permasalahan biologis tersebut mungkin saja dapat diselesaikan melalui eksperimen di laboratorium, tetapi tentu saja itu akan membutuhkan waktu dan sumber daya yang sangat banyak. Salah satu teknik modern untuk menyelesaikan permasalahan semacam ini adalah dengan menyelesaikannya secara komputasional menggunakan komputer. Namun, sebelum melakukannya, kita harus menerjemahkan permasalahan biologis tersebut ke permasalahan komputasional yang memang dapat diselesaikan oleh komputer secara algoritmis.
Pertama-tama, perlu diketahui bahwa permasalahan komputasional haruslah memiliki input dan expected output yang jelas. Apabila masih terdapat ambiguitas, maka kita tak dapat membuat langkah algoritma yang dapat dimengerti oleh komputer. Untuk itu, kita perlu terlebih dahulu memformulasikan input dan expected output-nya.
Dalam permasalahan ini, kita telah memiliki serentetan sekuens gen yang diaktivasi oleh DosR berdasarkan eksperimen penelitian sebelumnya, yang dalam artikel ini digunakan berjumlah 10. Karena protein faktor transkripsi DosR berikatan di daerah promoter dekat gen, kita dapat mengekstrak sebagian sekuens pada bagian promotor di setiap 10 gen tersebut. Di kasus ini, kita akan mengambil 250bp sekuens terdekat dengan gen. Jadi, input yang kita miliki adalah 10 untai DNA (direpresentasikan sebagai string pada komputer) dengan masing-masing string memiliki 250 huruf nukleotida.
Selanjutnya. kita perlu menentukan expected output-nya. Kita tahu dari sifat yang dimiliki DosR, yang pasti hanya dapat berikatan pada sekuens spesifik, dan dari situ kita bisa menyimpulkan bahwa: karena 10 gen itu sudah terbukti diregulasi oleh DosR, maka di bagian promoter masing-masing gen tersebut akan ada sekuens spesifik dengan panjang tertentu yang mirip antar satu sama lain. Kita dapat berasumsi bahwa tentulah terjadi mutasi pada masing-masing gen, tetapi kita tetap dapat berekspektasi bahwa untuk sekuens spesifik ini, mutasinya akan minim atau terkonservasi; karena jika tidak, sekuens-nya tidak akan terkenali oleh DosR yang berkontradiksi dengan fakta eksperimental kalau gen-nya diregulasi DosR. Atas dasar pengetahuan ini, kita dapat memformulasikan expected output yang kita inginkan, yaitu dalam bentuk sekuens spesifik yang hadir di masing-masing 10 gen tadi dan huruf dari sekuens tersebut harus memiliki perbedaan seminimal mungking.
Kita seharusnya dapat menemukan pesan tersembunyi dalam bentuk sekuens motif yang menjadi rahasia DosR dapat mengatur semua gen tersebut!
Input dan expected output kita sudah jelas. Oleh karena itu, permasalahan mencari pesan tersembunyi dari 10 gen tersebut dapat dirubah menjadi permasalahan komputasional sebagai berikut:
Permasalahan Pencarian Motif
Diberikan sekumpulan untai DNA dengan panjang , dan sebuah bilangan bulat , temukan sekumpulan -mer (satu dari setiap untai) yang meminimalkan skor perbedaan keseluruhan dari matriks motif yang dihasilkan.
Catatan: k-mer adalah istilah yang merujuk pada sekuens pendek DNA dengan panjang nukleotida
Beberapa Pendekatan Algoritma
Untuk menyelesaikan permasalahan pencarian motif tersebut, kita akan mencoba tiga pendekatan, yaitu brute force, heuristik, dan probabilistik. Tentu saja masing-masing algoritma akan memiliki kelemahan dan kekurangannya; dan itulah yang akan kita komparasikan satu sama lain di akhir nanti.
Strategi Brute Force
Pada strategi pencarian paksa (brute force), kita pada dasarnya membuat semua produk k-mers yang mungkin dari ukuran bilangan bulat k yang sudah ditentukan. Kemudian dari daftar semua k-mers tersebut, kita dapat mencocokan k-mers tersebut dengan setiap k-mers yang memang terdapat atau hadir di setiap sekuens DNA promoter, tentu saja dengan mempertimbangkan berapa banyak skor perbedaan yang ada di setiap kombinasinya. k-mers yang memiliki skor paling perbedaan paling minimal nanti yang akan menjadi hasil akhir sekuens motif-nya.
Namun, pendekatan brute force semacam itu sangatlah lambat, kita bisa coba perkirakan operational cost-nya. Ingat bahwa kita memiliki
buah untai DNA dengan masing-masingnya memiliki panjang
. Lalu kita mencari subsekuen atau k-mers dengan panjang
satu per satu. Oleh karena itu, kita akan memiliki search space (ruang pencarian) sebanyak
, dimana ekspresi
itu adalah kombinasi macam k-mers yang dapat dibentuk di satu untai DNA. Karena kita memiliki
, berarti kita memiliki search space sekita
atau setara dengan
. Angka ini begitu besar dan tidak realistis untuk input data yang kita miliki. Maka dari itu, untuk strategi brute force, supaya lebih realistis untuk dapat dilakukan, dilakukan pendekatan lain, yaitu Algoritma Median String 1
Median string sendiri adalah string representatif (consensus string) yang ditentukan dari kumpulan string tetapi yang meminimalisasi total skor perbedaan dari semua string yang ada pada kumpulan string tersebut. Nah, pendekatan algoritma ini akan memiliki search space yang jauh lebih kecil, meski tetap signifikan, dari pendekatan brute force manual seperti yang direpresentasikan pada Permasalahan Pencarian Motif di atas. Oke, langsunglah kita coba implementasikan.
Pertama-tama kita perlu memformulasikan skor perbedaan antar sekuens DNA secara matematis. Hal yang paling mudah untuk mengukur seberapa besar dua sekuens berbeda antar satu sama lain adalah dengan secara langsung menghitung jumlah perbedaan huruf di antara dua sekuens tersebut. Ukuran perbedaan ini disebut sebagai hamming_distance, yang apabila dituliskan dalam kode menjadi sebagai berikut:
def hamming_distance(seq1, seq2):
"""
Calculates the Hamming distance between two equal-length DNA sequences.
"""
# Hamming distance is only defined for sequences of the same length
if len(seq1) != len(seq2):
raise ValueError("Sequences must be of equal length.")
# Zip the sequences together and count mismatches
return sum(1 for base1, base2 in zip(seq1, seq2) if base1 != base2)
Kedua, kita perlu sebuah cara untuk menghasilkan semua sekuens k-mers yang mungkin ada untuk sebuah bilangan bilangan k. Jadi untuk , kita akan mempunyai total macam sekuens, untuk berarti ada (1 miliar) total sekuens dan seterusnya. Adapun untuk baris kode yang dapat menghasilkan k-mers tersebut adalah sebagai berikut:
def generate_kmers_lazy(k, alphabet="ACGT"):
"""Yields _k-mers_ one by one without storing them in memory."""
# itertools.product acts as a generator under the hood
for kmer_tuple in itertools.product(alphabet, repeat=k):
yield "".join(kmer_tuple)
Baris kode tersebut menghasilkan k-mers tanpa menyimpannya dalam memori (makanya nama fungsinya lazy). Hal ini perlu dilakukan karena apabila kita membuat fungsi terpisah untuk pembuatan k-mers ini dan hasilnya disimpan dalam sebuah variabel, nanti ukuran variabelnya bisa sangat besar. Akan lebih baik apabila kita mengimplementasikan fungsi ini secara langsung di algoritma utama tanpa tahapan penyimpanan di memori. Jadi ketika k-mers dihasilkan fungsi ini, k-mers akan langsung diproses pada tahapanan algoritma utama lebih lanjut.
Terakhir adalah algoritma utama dari Median String itu sendiri yang akan mencari k-mer spesifik yang memiliki skor perbedaan terkecil. Langkah algoritmanya sendiri adalah sebagai berikut:
- menghasilkan semua k-mers dari sebuah bilangan bulat k
- dari semua k-mers yang dibuat pada langkah pertama, semua kombinasi kumpulan k-mers disebut matrix motifs, dimana dalam matriks motifs tersebut akan terdapat k-mers dari masing-masing sekuens DNA promoter.
- Dari semua kombinasi matriks motifs tersebut, ditentukan mana kombinasi yang dapat meminimalkan skor total hamming distance dari masing-masing k-mers pada motif tersebut.
Adapun kode untuk implementasi algoritma ini adalah sebagai berikut:
def median_string(dna_strings, k):
"""
Finds a sequence motif in a collection of DNA strings
using the Median String algorithm approach.
"""
kmer_stream = generate_kmers_lazy(k)
consensus_kmer = ""
consensus_motifs = []
consensus_dist = inf
for kmer in kmer_stream:
best_motifs = []
best_motifs_dist = 0
# For each sequence, find the window that minimizes distance to the candidate kmer
for seq in dna_strings:
best_dist = inf
best_kmer = ""
# Slide a window of size k across the sequence
for i in range(len(seq) - k + 1):
window = seq[i : i + k]
ham_dist = hamming_distance(kmer, window)
if ham_dist < best_dist:
best_kmer = window
best_dist = ham_dist
best_motifs.append(best_kmer)
best_motifs_dist += best_dist
# Check if this candidate k-mer is the closest overall "median string"
if best_motifs_dist < consensus_dist:
consensus_kmer = kmer
consensus_motifs = best_motifs
consensus_dist = best_motifs_dist
return consensus_kmer, consensus_dist
Dari hasil akhir matriks motif terbaik yang meminimalkan total skor hamming distance, ditentukan juga consensus k-mer yang menjadi representasi utama dari sekuens motif yang dihipotesiskan berikatan langsung dengan DorS.
Dari algoritma utama yang telah dibuat, kita dapat memperkiraan computational cost yang diperlukan agar komputasi algoritma ini dapat berjalan. Tak perlu berpikir panjang, kita sebenarnya sudah dapat memperkirakan bahwa computational cost untuk algoriitma ini sangatlah besar, meski tak sebesar strategi brute force manual yang sebelumnya. Tapi, kita dapat memperkirakannya secara lebih presisi lagi, yaitu dengan mempertimbangkan berapa banyak operasi yang dibutuhkan pada masing-masing tahapan algoritma tersebut, yang kira-kira dapat disimpulkan melalui paragraf berikut:
Pertama, kita akan mendapati jumlah k-mers yang mungkin sebanyak . Kedua, dari semua k-mers yang terbentuk, kita mencari semua kombinasi k-mers yang mungkin sebanyak (dimana merujuk pada jumlah sekuens DNA promoter kita), berarti kombinasi totalnya ada . Terakhir, dari semua kombinasi tersebut, kita juga secara iteratif mencari bagian spesifik sekuens dari masing-masing DNA promoter yang memiliki kecocokan, dengan mempertimbangkan skor hamming distance yang minimal terhadap motif (dimana setiap kalkulasinya memerlukan jumlah operasi), sehingga total operasi yang diperlukan adalah . Dengan nilai dan , maka total operasi yang diperlukan adalah .
Algoritma Median String ini oleh karenanya memiliki search space
. Jika kita hanya menghitung
= 10, maka algoritma Algoritma Median String memiliki search space
.Angka ini meskipun sangat besar untuk nilai
besar, tetapi tidak semasif pendekatan brute force manual sebelumnya yang memiliki search space
.
Dari sini, kita dapat menyimpulkan bahwa melakukan strategi brute force apapun mesti memiliki skala eksponensial untuk operational cost-nya. Dan untuk algoritma yang kita implementasikan, dengan besar nilai lebih dari 12 itu membutuhkan waktu yang sangat lama dan tidak memungkinkan dijalankan menggunakan laptop (saya sudah mencobanya). Oleh karena itu, di sini saya membatasi eksekusi algoritma ini hanya untuk (yang ini memakan sekitar 1-2 jam).
Untuk mengeksekusinya dengan nilai yang berbeda-beda, kita tinggal membuat iterasi for loops saja:
for k in range(6,12):
motif = median_string(dna_strings)
print(f"k={k} | ", motif, "score: ", score)
Hasil dari eksekusi dari algoritma brute force sendiri adalah sebagai berikut:
| k | Motif | Score |
|---|---|---|
| 6 | ACGGCG | 5 |
| 7 | ACCGACG | 9 |
| 8 | CATCGGCC | 11 |
| 9 | ACCGACGGG | 16 |
| 10 | CTATCGGCCC | 19 |
| 11 | GGACTTCCGGC | 20 |
| 12 | GGACTTCCGGCC | 23 |
Pada dasarnya, kita tak tahu seberapa panjang sekuens spesifik (motif) yang memang dapat dikenali oleh DosR di kenyataannya. Ditambah lagi apabila kita mencermati hasil di atas, kita akan menemukan bahwa masing-masing nilai , menghasilkan consensus motif yang berbeda-beda. Ini memanglah membingungkan, tetapi itu sejatinya adalah hasil yang expected. Alasannya adalah di setiap nilai , kita memiliki ruang pencarian (search space) yang berbeda. Dari ruang pencarian yang berbeda itu tidak ada jaminan matematis bahwa hasil consensus string-nya memiliki kemiripan tinggi. Jadi bukannya algoritma brute force ini tidak akurat, justru ini adalah pendekatan yang paling akurat untuk setiap nilai .
Selain itu, algoritma strategi brute force juga sangat mahal secara kebutuhan komputasi. Hal ini yang menyebabkan ia jarang dipakai di dunia nyata karena praktikalitas kecepatannya yang tak memungkinkan untuk digunakan di mayoritas situasi.
Untuk strategi lebih cepat, kita akan lanjut ke pendekatan selanjutnya: strategi heuristik.
Strategi Heuristik (Algoritma Serakah)
Jika strategi brute force itu bodoh tetapi selalu akurat, maka padanan strategi yang agak lebih pintar adalah heuristic, yaitu sebuah strategi pendekatan jalan pintas untuk menyelesaikan masalah, meskipun terdapat harga yang harus dibayarkan, yaitu dalam bentuk akurasi yang berpotensi menurun.
Strategi heuristik yang akan digunakan di sini disebut greedy algorithm (algoritma serakah). Sebagai pengantar tentang greedy algorithm, kita bisa membayangkan satu analogi dalam bentuk permainan catur. Dalam permainan catur, di setiap langkah kita dihadapkan banyak sekali opsi dan untuk pemula opsi mana yang tepat tidaklah selalu jelas. Namun, jika pemula itu sudah diajarkan bahwa perwira itu bernilai lebih dibandingkan pion, serta perwira satu dengan perwira jenis lain bisa memiliki nilai yang berbeda (misalnya ratu dengan benteng). Dari sini, si pemula dapat menyusun strategi untuk menentukan pilihan pada setiap langkahnya, yaitu dengan mencari gerakan apa yang memaksimalkan reward. Jika di suatu langkah terdapat opsi memakan peluncur, benteng, dan pion; maka si pemula akan dapat secara otomatis memakan benteng. Nah, strategi semacam ini disebut sebagai algoritma serakah karena ia memaksimalkan reward di setiap tahapan langkah tanpa memperdulikan langkah sebelum dan setelahnya.
Strategi catur itu mungkin cukup baik untuk pemula, tetapi pemain catur yang cukup cakap akan langsung mengetahui kelemahan dari strategi ini. Untuk bermain catur dengan cakap, yang diperlukan tidak hanya mempertimbangkan reward terbesar di tahapan langkah saat ini, tetapi juga perlu melihat kombinasi kemungkinan yang muncul di langkah selanjutnya setelah gerakan dilakukan. Misalnya, jika musuh melakukan gerakan pengorbanan benteng untuk menyerang raja yang berpotensi skakmat, maka dengan strategi tadi si pemula akan langsung kalah.
Analogi catur ini cukup menggambarkn greedy algorithm dengan cukup baik. Pada dasarnya greedy algorithm adalah pendekatan penyelesaian masalah dengan cara membangun solusi dengan memilih opsi yang memaksimalkan reward pada tahapan saat ini tanpa mempertimbangkan tahapan setelah-setelahnya. Atau, dalam bahasa formal menjadi:
A greedy algorithm is an algorithm which, at each step, makes the choice that is locally optimal, and subsequently does not reconsider past choice.
Apabila pemahaman ini kita terapkan terhadap Pemasalah Pencarian Motif, kita dapat membayangkan masalahnya menjadi permasalahan mencari optimal k-mer di setiap sekuens DNA. Di masalah DosR, kita memiliki 10 sekuens DNA, berarti tugas greedy algorithm adalah menemukan k-mer optimal di setiap sekuens tersebut secara berurutan.
Untuk mengimplementasikan greedy algorithm, mula-mula kita perlu terlebih dahulu mendefinisikan apa yang dimaksud dengan optimal k-mer. Nah, untuk mendefinisikan ini, kita memerlukan satu konsep, yaitu motif profile matrix sekuens DNA.
Motif Profile Profil DNA didefinisikan sebagai matriks yang merepresentasikan distribusi probabilitas masing-masing nukeotida di setiap basa sekuens DNA. Sebagai ilustrasi, dapat diperhatikan gambar berikut:
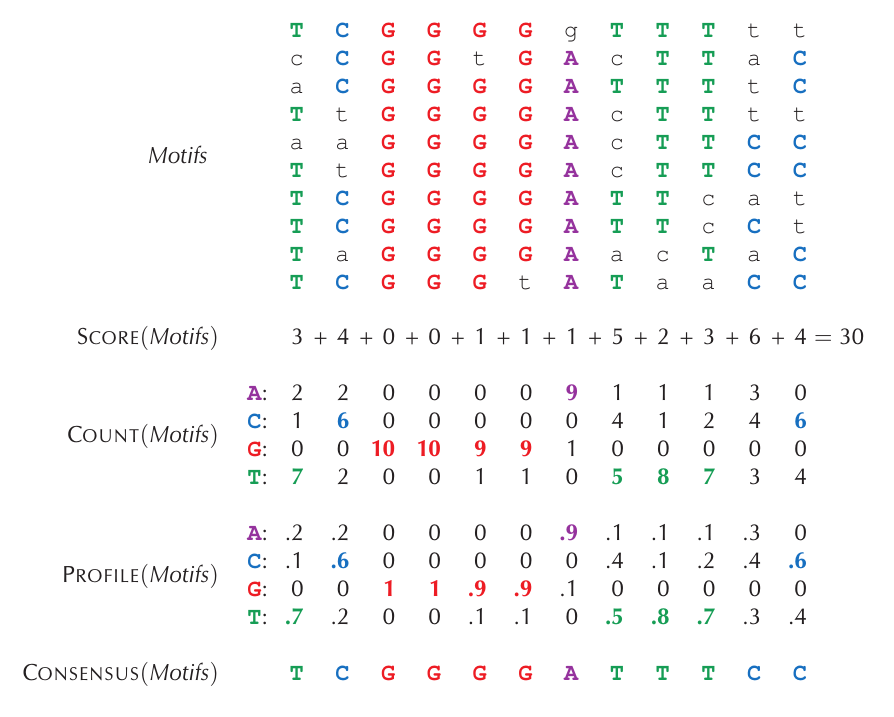
Bisa dilihat bahwa profile dihitung melalui frekuensi masing-masing jenis nukleotida di setiap posisi basa nukeotida pada kumpulan sekuens DNA. Dari profile ini kita dapat menentukan probabilitas kemunculan suatu sekuens k-mer, karena masing-masing posisi basa nukelotida sudah merepresentasikan probabilitas kemunculan masing-masing jenis nukelotida. Sebagai contoh, dapat dilihat gambar di bawah ini:

Apabila jeli, akan terlihat sebenarnya ada masalah dalam menghitung probabilitas berdasarkan profil frekuensi munrni seperti ini. Kalau saja salah satu dari posisi basa nukleotida ternyata memiliki nilai probabilitas
, maka probabilitas kemunculan sekuens penuh itu juga akan
. Memberikan nilai probabilitas 0 ke sebuah kemungkinan peristiwa, however unlikely, itu tidak diperbolehkan. Prinsip ini disebut sebagai Cromwell’s Rule, yang menyatakan bahwa kita tidak diperbolehkan memberikan kepastian (beri nilai probabilitas 0 dan 1) pada peristiwa yang sifatnya probabilistik. Oleh karena itu, kita perlu memodifikasi mekanisme pemberian probabilitas murni di atas agar nilainya tidak mungkin
mutlak. Hal ini bisa ditangani dengan menggunakan Laplace’s rule of succession, yaitu dengan menambah nilai
untuk nilai frekuensi nukelotida pada pembentukan profile matrix. Sebagai ilustrasi, bisa lihat contoh berikut:

Jadi, dengan dua hal tersebut, kita bisa mendefinisikan fungsi profile() dengan mempertimbangkan Laplace’s rule of succession dan prob() yang menentukan probabilitas kemunculan suatu sekuens kmer dari profile yang sudah ada.
def profile(motifs, k):
"""Generates a profile matrix using Laplace's Rule of Succession (Pseudocounts)."""
t = len(motifs)
# Start counts at 1.0 instead of 0.0
profile = {nucleotide: [1.0] * k for nucleotide in ['A', 'C', 'G', 'T']}
for col in range(k):
for motif in motifs:
nucleotide = motif[col]
profile[nucleotide][col] += 1
# Divide counts by (t + 4) because 4 virtual pseudocounts were added to each column
for nucleotide in profile:
for col in range(k):
profile[nucleotide][col] /= (t + 4)
def profile_most_probable_kmer(dna_string, k, profile):
"""Finds the Profile-most probable k-mer in a single DNA string."""
best_kmer = dna_string[0:k]
best_prob = -1
for i in range(len(dna_string) - k + 1):
window = dna_string[i:i+k]
prob = 1.0
for idx, nucleotide in enumerate(window):
prob *= profile[nucleotide][idx]
if prob > best_prob:
best_prob = prob
best_kmer = window
return best_kmer
Greedy Motif Search
Oke, memang tahapan konsep profile matrix dan profile-most-probable kmer tadi sangat panjang, tetapi sebenarnya apa gunanya untuk implementasi algoritma ini? Kunci untuk memahaminya adalah kita bisa menggunakan dua fungsi itu secara iteratif untuk membangun solusi motifs yang kita inginkan, yaitu motifs berisikan kumpulan sekuens k-mers yang memiliki total skor perbedaan hamming distance terkecil.
Jadi, dengan menggunakan fungsi profile() dan profile_most_probable_kmer() kita dapat menyelesaikan Permasalahan Pencarian Motif dengan membangun motifs satu k-mer per k-mer yang diambil di setiap sekuens DNA secara berurutan. Secara lebih lengkap, tahapan kerja algoritma ini adalah sebagai berikut:
- Kita membuat iterasi (outer loop) yang bergerak untuk setiap k-mer yang ada di sekuens pertama
Dna; k-mers di sini adalah yang menjadi sekuens motifs pertama (sebut sajamotifs[1]) - Untuk setiap k-mer pertama itu, kita menentukan profile matrix dengan Laplace’s Rule of Succession tadi
- Profile tersebut digunakan untuk k-mers
motifs[2]melalui fungsiprofile_most_probable_kmer() - Setelah
motif[2]ditentukan, kita membangun profile matrix lagi dengan menggunakan 2 sekuens sebelumnya dan profile matrix ini digunakan untuk menentukan sekuensmotifs[3]dan seterusnya - Langkah 2-4 dilakukan secara iteratif sampai sekuens terakhir, baru kemudian iterasi di langkah 1 berlanjut lagi untuk membangun
motifsbaru - Pada setiap
motifspenuh yang telah dibuat, kita menentukan total skor perbedaan hamming distance. Dari skor yang disimpan, kita akan menentukanmotifsmana yang memiliki skor minimal.
Untuk tahapan ke-6, kita dapat membuat fungsi baru score_motifs() yang pada dasarnya menghitung total hamming distance di setiap sekuens yang terdapat di dalamnya. Dari tahapan tersebut, kita dapat mengimplementasikannya dengan kode python berikut:
def score_motifs(motifs, k):
"""Calculates the total mismatch score for a collection of motifs."""
total_score = 0
t = len(motifs)
for col in range(k):
# Count occurrences of each base in this specific column
counts = {'A': 0, 'C': 0, 'G': 0, 'T': 0}
for motif in motifs:
counts[motif[col]] += 1
# Find the maximum count (the consensus nucleotide count)
max_count = max(counts.values())
# Mismatches in this column = total strings minus the consensus count
total_score += (t - max_count)
return total_score
def greedy_motif_search(dna_strings, k, t):
"""
Implements the Greedy Motif Search Algorithm.
dna_strings: List of t strings of DNA
k: Length of the motif
t: Number of strings in dna_strings
"""
# Initialize BestMotifs as the first k-mer from every sequence
best_motifs = [seq[0:k] for seq in dna_strings]
best_score = score_motifs(best_motifs, k)
# Grab the first string to find all potential 'seed' _k-mers_
first_seq = dna_strings[0]
# Loop over every possible k-mer in the first sequence
for i in range(len(first_seq) - k + 1):
# Step A: Start a brand new candidate motif collection with our seed
current_motifs = [first_seq[i:i+k]]
# Step B: Iteratively evaluate the remaining sequences (from index 1 to t-1)
for j in range(1, t):
# 1. Create a profile based ONLY on the motifs collected so far
current_profile = priofile(current_motifs, k)
# 2. Find the most probable k-mer in the j-th sequence using that profile
next_best_kmer = most_probable_kmer(dna_strings[j], k, current_profile)
# 3. Add it to our growing candidate collection
current_motifs.append(next_best_kmer)
# Step C: Compare our fully built collection to our all-time best
current_score = score_motifs(current_motifs, k)
if current_score < best_score:
best_score = current_score
best_motifs = current_motifs
return best_motifs
Eksekusi dan Hasil
Seperti sebelumnya, kita dapat menjalankan algoritma untuk beberapa nilai k sekaligus. Namun, karena algoritma ini terhitung cepat, akan dilakukan sampai nilai
.
| k | motif | score |
|---|---|---|
| 6 | CGCCGG | 6 |
| 7 | CGGCCAG | 9 |
| 8 | CCGGCGGG | 12 |
| 9 | TATCGGCCC | 17 |
| 10 | CTATCGGCCC | 19 |
| 11 | GGACTTCCGGC | 20 |
| 12 | GGACTTCCGGCC | 24 |
| 13 | GGACTTCCGGCCC | 28 |
| 14 | GGGACTTCCGGCCC | 33 |
| 15 | GGACTTACGGCCCTA | 35 |
| 16 | GGACTAACGACCCTAG | 40 |
| 17 | GGGACCAACGACCCTAG | 44 |
| 18 | GGGGACCAACGACCCTAG | 49 |
| 19 | GGGGACCAACGACCCTAGC | 55 |
| 20 | GGGGACCTACGTCCCTAGCC | 56 |
Algoritma greedy ini, jika dibandingkan brute force melalui Algprotma Median String sebeleumnya adalah langit dan bumi secara kecepatan. Bahkan, untuk menjalankan keseluruhan algoritma untuk nilai
bervarisai (6-20), hanya membutuhkan sekitar 1-5 menit untuk selesai. Sangat berbeda dengan sebelumnya yang bahkan untuk menjalankan algoritma untuk
bisa memakan waktu setengah jam lebih.
Hal ini disebabkan karena memang operation cost dari algoritma ini terhitung rendah, yang dapat kita aproksimasi dengan menghitung jumlah search space yang digunakan untuk menyelesaikan algoritma ini. Pertama, algoritma serakah ini mencari setiap k-mers di sekuens pertama, yang berjumlah
, dengan
adalah panjang sekuens DNA (dalam konteks data ini berarti 250bp). Kedua, dari setiap iterasi k-mers dilakukan iterasi lebih lanjut untuk setiap sekuens selanjutnya (berjumlah
), di mana di setiap sekuens tersebut dijalankan fungsi profle() dan profile_most_probable_kmer() yang memiliki operation cost sekitar
dan
secara berurutan. Menggabungkan semuanya kita dapat mengestimasi bahwa operation cost keseluruhan algoritma ini hanyalah
, yang jauh lebih kecil dibandingkan sebelumnya yang memerlukan
.
Meskipun cepat, kita dapat melihat sedikit penurunan akurasi dari hasil consensus motif yang dihasilkan dari algoritma ini, yaitu melalui peningkatan skor total perbedaan hamming distance secara konsisten dibandingkan dengan algoritma Median String sebelumnya. Kelemahan lain yang cukup signifikan dari algoritma ini adalah ketergantungan besarnya terhadap k-mers yang digunakan sebagai seed di sekuens pertama, yang digunakan untuk membuat profile matrix pertama kalinya. Jika saja kita memiliki dataset berjumlah banyak sekuens, tapi kebetulan sekuens pertama dalam dataset tidak mengandung motif yang kita cari, maka dengan menggunakan algoritma ini kita tak akan pernah mendapatkan kumpulan motifs yang kita inginkan.
Terlepas dari itu, greedy algorithm tetaplah menjadi salah satu tools dalam arsenal peneliti computational biology karena efisiensi komputasinya membuat algoritma ini dapat diaplikasikan untuk data yang besar.
Strategi Probabilistik (Algoritma Monte Carlo)
Jika brute force terlalu lambat, tetapi heuristic terlalu serakah dan bodoh, Bisakah kita mendapatkan the best of both world? Jawabannya adalah iya, meskipun tidak sepenuhnya. Hal ini dilakukan dengan menggunakan pendekatan yang sepenuhnya berbeda dari sebelum-sebelumnya, yaitu dengan memanfaatkan randomness atau keacakan. Memang aneh kalau dipikirkan secara sekilas. Bagaimana algoritma yang bergantung pada keacakan bisa menghasilkan solusi terhadap permasalahan. Tapi memang begitulah kenyataannya; banyak benda di alam semesta (bintang, kehidupan, dll) ini pun juga bisa dipikirkan sebagai keteraturan yang muncul dari kecenderungan alam semesta untuk menuju ke ketidakteraturan (entropy), yang dengan kata lain ada pergerakan dari keacakan menjadi ketidakacakan, but I digress.
Randomized Motif Search
Kembali ke topik utama, bagaimana algoritma probabilstik ini bekerja? Ingat sebelumnya kita memiliki fungsi profile(motifs) yang dapat menghasilkan profile matrix dari motifs (sekumpulan sekuens k-mers). Sekarang, dari kumpulan strings sekuen Dna dan sebuah profile matrix, kita akan mendefinisikan fungsi motifs(profile, Dna) yang akan menghasilkan kumpulan k-mers yang masing-masingnya berasal dari hasil fungsi profile_most_probable_kmer() yang dijalankan pada setiap sekuen DNA. Sebagai ilustrasi, bisa di lihat pada gambar berikut.

Nah, kunci dari algoritma ini adalah dengan secara terus-menerus mengulang proses pembentukan motifs menggunakan fungsimotifs()dan mengupdate profile matrix di setiap motifs yang terbentuk pada setiap iterasi. Dengan kata lain, kita terus mengiterasi rentetan fungsi berikut:
Iterasi secara terus menerus dilakukan selama skor total perbedaan hamming distance dari motifs yang terbentuk terus menurun (kualitas motifs-nya meningkat terus). Jadi, secara kasar kita dapat menuliskan algoritmanya menjadi beberapa tahapan berikut:
- membuat motifs awal dengan secara acak memilih k-mers pada setiap sekuen DNA sebagai langkah inisiasi
- membuat while loop yang akan terus berjalan untuk membuat profile matrix menggunakan fungsi
profile(motifs)terhadap motifs yang dibuat di iterasi sebelumnya dan membentuk motifs baru dengan menjalankan fungsimotifs - langkah iterasi di tahapan 2 berjalan secara terus menerus kecuali sampai pada kondisi dimana skor hamming distance yang dihasilkan pada iterasi terakhir lebih kecil daripada yang dihasilkan pada iterasi sebelunya.
Tahapan algoritma ini apabila diimplementasikan dalam kode python akan terlihat demikian:
import random
def randomized_motif_search(dna_strings, k, t):
"""
Runs one instance of the Randomized Motif Search algorithm.
"""
# 1. Randomly select initial motifs uniformly
motifs = []
for dna in dna_strings:
random_idx = random.randint(0, len(dna) - k)
motifs.append(dna[random_idx : random_idx + k])
best_motifs = motifs
while True:
# 2. Build profile with pseudocounts from current motifs
profile = profile(motifs)
# 3. Form new motifs by finding the Profile-most Probable k-mer in each string
motifs = [profile_most_probable_kmer(dna, k, profile) for dna in dna_strings]
# 4. Compare scores
if score(motifs) < score_motifs(best_motifs):
best_motifs = motifs
else:
return best_motifs
def repeated_randomized_motif_search(dna_strings, k, t, iterations=1000):
"""
Runs RandomizedMotifSearch multiple times to avoid local optima
and returns the best motifs found across all runs.
"""
# 1. Run the algorithm once to establish a baseline "best"
best_motifs = randomized_motif_search(dna_strings, k, t)
best_score = score_motifs(best_motifs)
# 2. Run the algorithm 999 more times
for i in range(1, iterations):
# Get the final motifs from a fresh random start
current_motifs = randomized_motif_search(dna_strings, k, t)
current_score = score_motifs(current_motifs)
# 3. Compare with our all-time best
if current_score < best_score:
best_motifs = current_motifs
best_score = current_score
return best_motifs
Memang, apabila kita menjalakan algoritma ini satu kali, kita kemungkinan besar akan mendapatkan hasil yang buruk. Tapi, jika kita menjalankan algoritma ini sebanyak 1000 kali atau lebih dan menyimpan motifs yang memiliki skor terbaik, maka dapat dipastikan kita akan mendapatkan hasil yang cukup baik.
Hasil Randomized Algorithm
Setelah menjalankan algoritma untuk mencari motif dengan ini sebanyak 1000 kali, didapatkan hasil berikut:
| K | Sequence | Score |
|---|---|---|
| 6 | ACGGCG | 5 |
| 7 | CTTCGGC | 9 |
| 8 | CTTCGGCC | 11 |
| 9 | GGCGGGGAC | 16 |
| 10 | ATCGACCCCA | 19 |
| 11 | GACCATCGGCC | 20 |
| 12 | GACCTACGGCCC | 24 |
| 13 | GGACCTACGGCCC | 28 |
| 14 | GGACTTACGGCCCT | 31 |
| 15 | GGACTAACGGCCCTA | 35 |
| 16 | CGGGACCTACGTCCCT | 39 |
| 17 | CGGGACCTACGTCCCTA | 43 |
| 18 | GGGACCTACGTCCCTAGC | 46 |
| 19 | GGGACCTACGGCCCTAGCC | 50 |
| 20 | CGGGACCTACGTCCCTAGCC | 55 |
Hasil dari Randomized Algorithm di atas memiliki skor yang konsisten lebih baik (lebih rendah) dibandingkan hasil greedy algorithm, meskipun untuk waktu eksekusinya lebih lambat. Tetapi, apabila sedikit jeli, akan terlihat di beberapa hasil, Randomized Algorithm memiliki nilai skor lebih tinggi. Hal ini tak terhindarkan karena memang nature dari algoritma ini yang probabilistik, sehingga hasil running bisa berbeda-beda; hasilnya seringkali lebih baik daripada greedy algorithm tetapi terkadang tidak. Namun, keuntungan dari algoritma ini adalah kita bisa membuat hasilnya memiliki skor lebih rendah lagi apabila kita melakukan iterasi lebih banyak lagi, misalkan 10000; meskipun dengan catatan bahwa waktu eksekusinya akan lebih lambat lagi.
Jika dibandingkan dengan Median String, memang algoritma ini kalah secara akurasi. Namun, sebenarnya bedanya tidak jauh dan kecepatan algoritma ini jauh lebih cepat dan bisa dijalankan untuk nilai yang besar sekaligus. Oleh karena itu, pendekatan algoritma ini sangat versatile untuk berbagai macam kasus permasalahan pencarian motif.
Komparasi Hasil dari Berbagai Algoritma dan Sekuens Motif Sebenarnya
Setelah kita jalankan ketiga algoritma yang dibuat, tahapan terakhir adalah membandingkan dengan sekuens motif binding site dari DosR yang sebenarnya. Kita dapat mendapatkan sekuens motif sebenarnya dari hasil penelitian dan eksperimen sebelumnya, salah satunya dari paper berikut: Park et al. (2007). Dalam eksperimen tersebut, sekuen motifnya adalah sekuen 20bp dalam bentuk palindrom2 5' TTSGGGACTWWAGTCCCSAA 3' (dengan W=C/G dan S=A/T). Namun, untuk mensimplifikasinya, sekuens binding site yang akan dipergunakan adalah sekuen 15bp: GGGACTTTGGGTACT; dengan mengambil bagian inti sekuen yang terkonservasi. Sekuen ini yang juga digunakan dalam buku Bioinformatics Algorithm dan juga akan digunakan sebagai ground truth dalam artikel ini. Tentu saja, untuk mengkomparasikan dengan hasil algoritma, kita akan memilih hasil dari nilai
yang sesuai dengan panjang sekuen sebenarnya tersebut, yaitu 15. Namun, karena hasil Median String tidak sampai
, maka kita akan menggunakan yang nilai
karena itu yang terbesar.
Perbandingan Hasil
Jika hasil motif dari masing-masing algoritma dikomparasikan dengan sekuen target 15bp tadi, kita akan mendapatkan alignment sebagai berikut.
| Algorithm | Motif Alignment | Overlap Similarity |
|---|---|---|
| Brute Force (Median String) |
GGGACTTTGGGTACT-llllll--ll---GGACTTCCGGCC |
72.73% (8/11 matches) |
| Heuristic (Greedy Motif Search) |
GGGACTTTGGGTACT-llll----l---l-GGACCGAAGTCCCCG |
42.86% (6/14 matches) |
| Probabilistic (Randomized Motif Search) |
GGGACTTTGGGTACT-llllll--ll--ll-GGACTTACGGCCCTA |
71.43% (10/14 matches) |
Dari hasil tersebut, kita dapat mengetahui bahwa strategi brute force memang yang terbaik karena algoritma ini menjamin mendapatkan kumpulan k-mer yang memiliki total skor perbedaan terkecil. Di sisi lain, strategi heuristic melalui Greedy Motif Search gagal total. Hal ini disebabkan karena sekuens bakteri TBC sendiri memiliki karakteristik unik, yaitu nilai GC-content-nya yang sangat tinggi, spesifiknya . Alhasil, algoritma greedy yang mengambil opsi dengan reward lokal maksimal di awal dan algoritmanya juga tidak mempertimbangkan informasi di langkah sebelumnya, membuat ia mudah terjebak dalam local optimum yang salah. Dalam hal ini, local optimum yang salah adalah daerah dengan rentetan G-C panjang.
Di sisi lain, hasil Randomized Motif Search mendekati akurasi dari brute force, dan dengan tambahan kelebihan komputasinya yang efisien. Hal ini disebabkan karena algoritma ini melakukan strategi yang memanfaatkan probabilitas dan dilakukan dalam pengulangan/iterasi yang banyak. Kita bisa membayangkan bahwa dari 250bp masing-masing sekuens DNA, mayoritas darinya memang noise, tetapi terdapat daerah tertentu yang memang penuh signal (ditandai dengan profile matrix yang berbeda karakter dengan noise background). Sebagian besar dari 1.000 running tersebut memang akan terjebak di zona kaya G-C yang salah. Namun, karena posisi awal diacak, ada beberapa running beruntung yang jendela awalnya tepat jatuh di atas sekuens DosR yang asli. Sekali matriks probabilitas (Profile) menyentuh sinyal DosR asli, ia bertindak seperti magnet yang menarik sekuens dari baris-baris lain keluar dari jebakan background noise.
Konklusi
Dari keseluruhan pembahasan yang telah dibuat, dapat disimpulkan bahwa permasalahan biologis dapat diselesaikan secara komputasional, tetapi dengan catatan bahwa permasalahan biologis tersebut harus dapat direpresentasikan dengan baik dalam komputer. Algoritma komputer yang tak mengetahui apa-apa tentang pengetahuan protein, gen, atau bakteri, secara independen dapat mendeteksi keberadaan pesan rahasia yang memiliki nilai informasi tinggi, seperti pada kasus pencarian motif ini.
-
Untuk pembuktian kenapa algoritma Median String bisa ekuivalen dengan pendekatan algoritma awal (pencocokan k-mers di setiap sekuens satu per satu), bisa langsung saja lihat di buku Bioinformatics Algorithm halaman 79-82. ↩︎
-
Palindrom adalah istilah untuk menyebut sekuen yang apabila dibaca dari depan atau belakang menghasilkan sekuen yang sama. Contohnya: “ATCCTA” jika dibalik “ATCCTA”. ↩︎